
Маркеры ДНК, связанные с сайтом рестрикции - Restriction site associated DNA markers
Маркеры ДНК, связанные с сайтом рестрикции (RAD), представляют собой тип генетического маркера, который полезен для ассоциативного картирования, QTL-картирования , популяционной генетики , экологической генетики и эволюционной генетики. Использование маркеров RAD для генетического картирования часто называют картированием RAD. Важным аспектом маркеров и картирования RAD является процесс выделения тегов RAD, которые представляют собой последовательности ДНК, которые непосредственно фланкируют каждый экземпляр определенного сайта рестрикции рестрикционного фермента по всему геному. После выделения меток RAD их можно использовать для идентификации и генотипирования полиморфизмов последовательностей ДНК, главным образом в форме однонуклеотидных полиморфизмов (SNP) . Полиморфизмы, которые идентифицируются и генотипируются путем выделения и анализа тегов RAD, называются маркерами RAD.
Изоляция тегов RAD
Использование фланкирующих последовательностей ДНК вокруг каждого сайта рестрикции является важным аспектом тегов RAD. Плотность меток RAD в геноме зависит от рестрикционного фермента, используемого в процессе выделения. Существуют и другие методы маркеров сайта рестрикции, такие как RFLP или полиморфизм длины амплифицированного фрагмента (AFLP), в которых используется полиморфизм длины фрагмента, вызванный разными сайтами рестрикции, для различения генетического полиморфизма. Использование фланкирующих ДНК-последовательностей в методах RAD-меток называется методом уменьшенного представления.
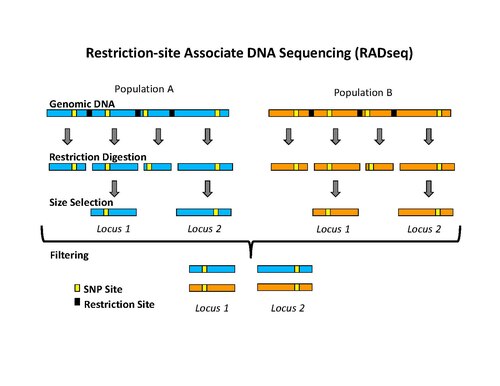
Процедура начальная , чтобы изолировать тег RAD участвует переваривание ДНК с определенным ферментом рестрикции, лигирование биотинилированного адаптеры к свесам, случайным образом стрижки ДНК на фрагменты гораздо меньше , чем среднее расстояние между сайтами рестрикции, и выделением биотинилированных фрагментов с использованием стрептавидином шариков. Первоначально эта процедура использовалась для выделения меток RAD для анализа микрочипов . Совсем недавно процедура изоляции тегов RAD была модифицирована для использования с высокопроизводительным секвенированием на платформе Illumina , что дает преимущество в виде значительного снижения количества необработанных ошибок и высокой пропускной способности. Новая процедура включает переваривание ДНК с помощью определенного рестрикционного фермента (например: SbfI, NsiI,…), лигирование первого адаптера, называемого P1, к выступам, случайным образом разрезая ДНК на фрагменты, намного меньшие, чем среднее расстояние между сайтами рестрикции. подготовка срезанных концов к тупым концам и лигирование второго адаптера (P2), а также использование ПЦР для специфической амплификации фрагментов, содержащих оба адаптера. Важно отметить, что первый адаптер содержит штрих-код короткой последовательности ДНК, называемый MID (молекулярный идентификатор), который используется в качестве маркера для идентификации различных образцов ДНК, которые объединены вместе и секвенированы в одной реакции. Использование высокопроизводительного секвенирования для анализа тегов RAD можно классифицировать как секвенирование с сокращенным представлением, которое включает, среди прочего, RADSeq (RAD-Sequencing).
Обнаружение и генотипирование маркеров RAD
После выделения меток RAD их можно использовать для идентификации и генотипирования полиморфизмов последовательностей ДНК, таких как однонуклеотидные полиморфизмы (SNP). Эти полиморфные сайты называют маркерами RAD. Самый эффективный способ найти теги RAD - это высокопроизводительное секвенирование ДНК , которое называется секвенированием тегов RAD, секвенированием RAD, RAD-Seq или RADSeq.
До разработки технологий высокопроизводительного секвенирования маркеры RAD были идентифицированы путем гибридизации тегов RAD с микрочипами. Из-за низкой чувствительности микрочипов этот подход может обнаруживать только полиморфизмы последовательностей ДНК, которые нарушают сайты рестрикции и приводят к отсутствию меток RAD, либо существенные полиморфизмы последовательностей ДНК, которые нарушают гибридизацию меток RAD. Следовательно, плотность генетических маркеров, которая может быть достигнута с помощью микрочипов, намного ниже, чем это возможно с помощью высокопроизводительного секвенирования ДНК.
История
Маркеры RAD были сначала реализованы с использованием микрочипов, а затем адаптированы для NGS (Next-Generation-Sequencing). Он был разработан совместно лабораториями Эрика Джонсона и Уильяма Креско в Университете Орегона примерно в 2006 году. Они подтвердили полезность маркеров RAD, определив контрольные точки рекомбинации у D. melanogaster и обнаружив QTL у трехиглой колюшки.
ddRADseq
В 2012 году был предложен модифицированный метод тегирования RAD под названием двойной дайджест RADseq (ddRADseq). Путем добавления второго рестрикционного фермента, замены случайного сдвига и жесткого выбора размера ДНК можно выполнить недорогое генотипирование популяции. Это может быть особенно мощным инструментом для сканирования всего генома для отбора и дифференциации популяции или адаптации популяции.